Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK
The major energy source for most cells is glucose, from which ATPis generated via glycolysis and/or oxidative metabolism. Glucosedeprivation activates AMP-activated protein kinase (AMPK)1, but itis unclear whether this activation occurs solely via changes in AMPor ADP, the classical activators of AMPK2–5. Here, we describe anAMP/ADP-independent mechanism that triggers AMPK activationby sensing the absence of fructose-1,6-bisphosphate (FBP), withAMPK being progressively activated as extracellular glucose andintracellular FBP decrease. When unoccupied by FBP, aldolasespromote the formation of a lysosomal complex containing at leastv-ATPase, ragulator, axin, liver kinase B1 (LKB1) and AMPK, whichhas previously been shown to be required for AMPK activation6,7.Knockdown of aldolases activates AMPK even in cells with abundantglucose, whereas the catalysis-defective D34S aldolase mutant, whichstill binds FBP, blocks AMPK activation. Cell-free reconstitutionassays show that addition of FBP disrupts the association of axinand LKB1 with v-ATPase and ragulator. Importantly, in some celltypes AMP/ATP and ADP/ATP ratios remain unchanged duringacute glucose starvation, and intact AMP-binding sites on AMPKare not required for AMPK activation. These results establish thataldolase, as well as being a glycolytic enzyme, is a sensor of glucoseavailability that regulates AMPK.
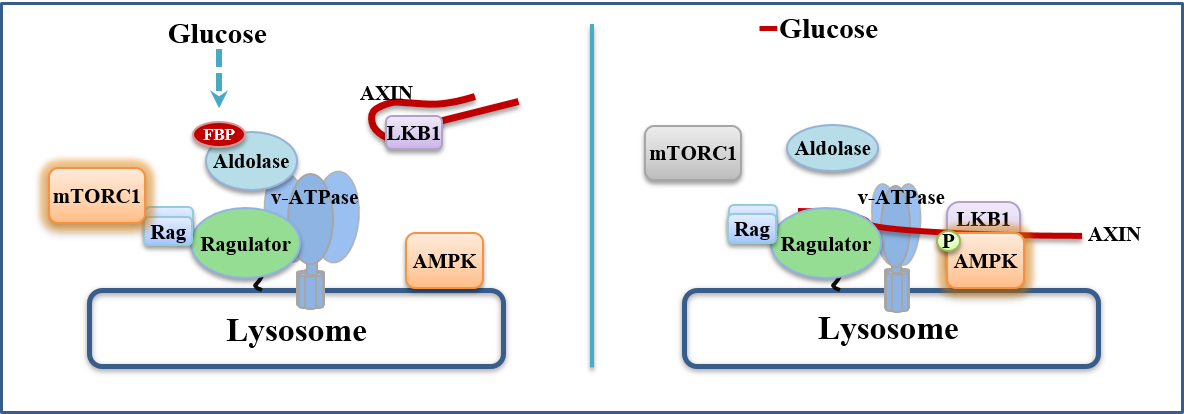
Zhang CS et al., Nature, 548(7665), 112-116, 2017
ULK1/2 Constitute a Bifurcate Node Controlling Glucose Metabolic Fluxes in Addition to Autophagy
Metabolic reprogramming is fundamental to biological homeostasis, enabling cells to adjust metabolic routes after sensing altered availability of fuels and growth factors. ULK1 and ULK2 represent key integrators that relay metabolic stress signals to the autophagy machinery. Here, we demonstrate that, during deprivation of amino acid and growth factors, ULK1/2 directly phosphorylate key glycolytic enzymes including hexokinase (HK), phosphofructokinase 1 (PFK1), enolase 1 (ENO1), and the gluconeogenic enzyme fructose-1,6-bisphosphatase (FBP1). Phosphorylation of these enzymes leads to enhanced HK activity to sustain glucose uptake but reduced activity of FBP1 to block the gluconeogenic route and reduced activity of PFK1 and ENO1 to moderate drop of glucose-6-phosphate and to repartition more carbon flux to pentose phosphate pathway (PPP), maintaining cellular energy and redox homeostasis at cellular and organismal levels. These results identify ULK1/2 as a bifurcate-signaling node that sustains glucose metabolic fluxes besides initiation of autophagy in response to nutritional deprivation.
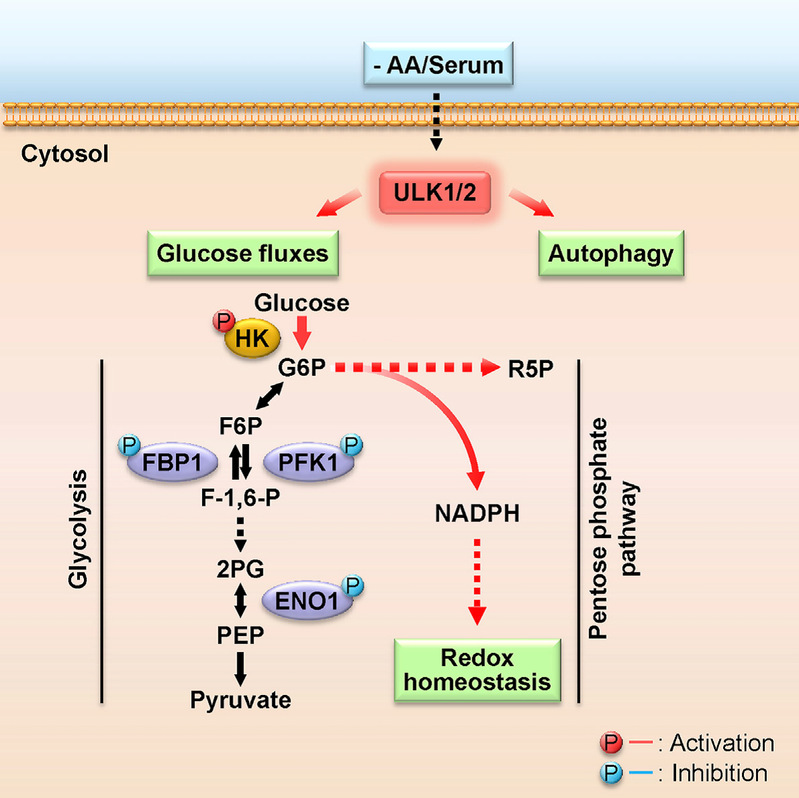
Li et al., Molecular Cell, 62, 359–370, 2016
The Lysosomal v-ATPase-Ragulator Complex is a Common Activator for AMPK and mTORC1, acting as a Switch between Catabolism and Anabolism
AMPK and mTOR play principal roles in governing metabolic programs; however, mechanisms underlying the coordination of the two inversely regulated kinases remain unclear. In this study we found, most surprisingly, that the late endosomal/lysosomal protein complex v-ATPase-Ragulator, essential for activation of mTORC1, is also required for AMPK activation. We also uncovered that AMPK is a residential protein of late endosome/lysosome. Under glucose starvation, the v-ATPase-Ragulator complex is accessible to AXIN/LKB1 for AMPK activation. Concurrently, the guanine nucleotide exchange factor (GEF) activity of Ragulator toward RAG is inhibited by AXIN, causing dissociation from endosome and inactivation of mTORC1. We have thus revealed that the v-ATPase-Ragulator complex is also an initiating sensor for energy stress and meanwhile serves as an endosomal docking site for LKB1-mediated AMPK activation by forming the v-ATPase-Ragulator-AXIN/LKB1-AMPK complex, thereby providing a switch between catabolism and anabolism. Our current study also emphasizes a general role of late endosome/lysosome in controlling metabolic programs.
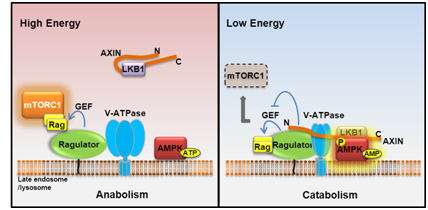
Zhang et al., Cell Metabolism, 20, 526-540, 2014
AMP as a low energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation
The AMP-activated protein kinase (AMPK) is a master regulator of metabolic homeostasis by sensing cellular energy status. AMPK is mainly activated via phosphorylation by LKB1 when cellular AMP/ADP levels are increased. However, how AMP/ADP brings about AMPK phosphorylation remains unclear. Here, we show that it is AMP, but not ADP, that drives AXIN to directly tether LKB1 to phosphorylate AMPK. The complex formation of AXIN-AMPK-LKB1 is greatly enhanced in glucose-starved or AICAR-treated cells and in cell-free systems supplemented with exogenous AMP. Depletion of AXIN abrogated starvation-induced AMPK-LKB1 colocalization. Importantly, adenovirus-based knockdown of AXIN in the mouse liver impaired AMPK activation and caused exacerbated fatty liver after starvation, underscoring an essential role of AXIN in AMPK activation. These findings demonstrate an initiating role of AMP and demonstrate that AXIN directly transmits AMP binding of AMPK to its activation by LKB1, uncovering the mechanistic route for AMP to elicit AMPK activation by LKB1.
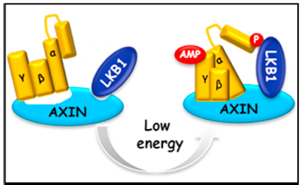
Zhang et al., Cell Metabolism, 18, 546–555, 2013
GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy
In metazoans, cells depend on extracellular growth factors for energy homeostasis. We found that glycogen synthase kinase-3 (GSK3), when deinhibited by default in cells deprived of growth factors, activates acetyltransferase TIP60 through phosphorylating TIP60-Ser(86), which directly acetylates and stimulates the protein kinase ULK1, which is required for autophagy. Cells engineered to express TIP60(S86A) that cannot be phosphorylated by GSK3 could not undergo serum deprivation-induced autophagy. An acetylation-defective mutant of ULK1 failed to rescue autophagy in ULK1(-/-) mouse embryonic fibroblasts. Cells used signaling from GSK3 to TIP60 and ULK1 to regulate autophagy when deprived of serum but not glucose. These findings uncover an activating pathway that integrates protein phosphorylation and acetylation to connect growth factor deprivation to autophagy.

Lin et al., Science, 336, 477, 2012.
Axin determines cell fate by controlling the p53 activation threshold after DNA damage
Cells can undergo either cell-cycle arrest or apoptosis after genotoxic stress, based on p53 activity. Here we show that cellular fate commitment depends on Axin forming distinct complexes with Pirh2, Tip60, HIPK2 and p53. In cells treated with sublethal doses of ultra-violet (UV) radiation or doxorubicin (Dox), Pirh2 abrogates Axin-induced p53 phosphorylation at Ser 46 catalysed by HIPK2, by competing with HIPK2 for binding to Axin. However, on lethal treatment, Tip60 interacts with Axin and abrogates Pirh2-Axin binding, forming an Axin-Tip60-HIPK2-p53 complex that allows maximal p53 activation to trigger apoptosis. We also provide evidence that the ATM/ATR pathway mediates the Axin-Tip60 complex assembly. An axin mutation promotes carcinogenesis in Axin(Fu)/+ (Axin-Fused) mice, consistent with a dominantnegative role for Axin(Fu) in p53 activation. Thus, Axin is a critical determinant in p53-dependent tumour suppression in which Pirh2 and Tip60 have different roles in triggering cell-cycle arrest or apoptosis depending on the severity of genotoxic stress.
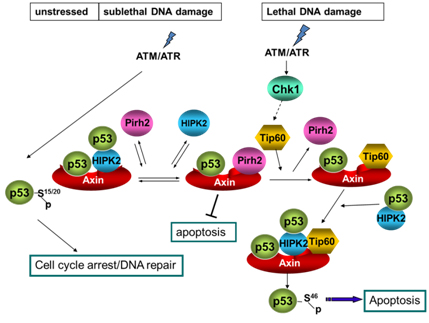
Li et al., Nature Cell Biology, 11, 1128-1135, 2009.
Axin stimulates p53 functions by activation of HIPK2 kinase through multimeric complex formation
Axin and p53 are tumor suppressors, controlling cell growth, apoptosis, and development. We show that Axin interacts with homeodomain-interacting protein kinase-2 (HIPK2), which is linked to UV-induced p53-dependent apoptosis by interacting with, and phosphorylating Ser 46 of, p53. In addition to association with p53 via HIPK2, Axin contains a separate domain that directly interacts with p53 at their physiological concentrations. Axin stimulates p53-dependent reporter transcription in 293 cells, but not in 293T, H1299, or SaOS-2 cells that are defective in p53 signaling. Axin, but not AxindeltaHIPK2, activates HIPK2-mediated p53 phosphorylation at Ser 46, facilitating p53-dependent transcriptional activity and apoptosis. Specific knockdown of Axin by siRNA reduced UV-induced Ser-46 phosphorylation and apoptosis. Kinase-dead HIPK2 reduced Axin-induced p53-dependent transcriptional activity, indicating that Axin stimulates p53 function through HIPK2 kinase activity. Interestingly, HIPK2deltaAxin that lacks its Axin-binding region acts as a dominant-positive form in p53 activation, suggesting that the Axin-binding region of HIPK2 is a putative autoinhibitory domain. These results show that Axin acts as a tumor suppressor by facilitating p53 function through integration of multiple factors.
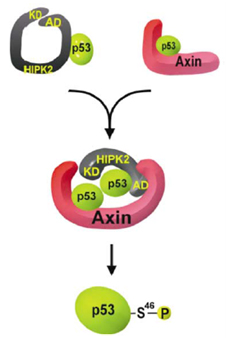
Rui et al., EMBO J., 23, 4583-4594, 2004